Lack of mitochondria causes severe disease in children

Researchers at Karolinska Institutet have discovered that excessive degradation of the power plants of our cells plays an important role in the onset of mitochondrial disease in children. These inherited metabolic disorders can have severe consequences such as brain dysfunction and neurological impairment. The study is published in EMBO Molecular Medicine.

“This is a completely new disease mechanism for mitochondrial disease which may provide a novel entry point for treating affected patients,” says Nils-Göran Larsson, professor at the Department of Medical Biochemistry and Biophysics, Karolinska Institutet, who led the study.
Mitochondrial diseases are inherited metabolic disorders that affect about 1 in 4,300 individuals and are caused by dysfunctional mitochondria. Mitochondria are the power plants of our cells and are crucial for converting energy derived from our food into the energy currency that drives the cell’s biochemical functions. Not surprisingly, organs that are mainly affected in patients are those with a high energy demand, such as the brain, heart, skeletal muscles, eyes and ears. In children, severe multisystem involvement and neurodegeneration are frequent manifestations.
Severe consequences
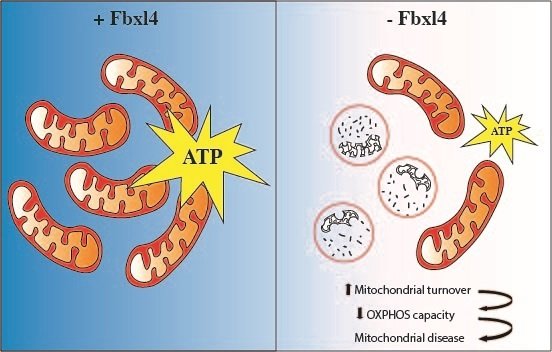
FBXL4 is a gene that is implicated in controlling mitochondrial function, and mutations in this gene are one of the most common causes of mitochondrial diseases. FBXL4 mutations have been linked to encephalopathy, a form of brain dysfunction causing neurological impairment. The manifestations are impaired cognitive function, developmental regression, epileptic seizures and other types of neurological deficits. Despite the severe consequences of FBXL4 mutations in humans, the function of the protein that FBXL4 codes for has remained poorly understood.
In the current study, researchers generated mice that lack FBXL4 and showed that these mice recapitulate important characteristics present in patients with FBXL4 mutations. They were able to demonstrate that the reduced mitochondrial function is caused by increased degradation of mitochondria via a process called autophagy.
Too few mitochondria in the tissues
In the absence of FBXL4, mitochondria are more frequently delivered to the lysosome, the recycling station of the cell that contains enzymes that break down organic compounds. FBXL4 thus acts as a break on mitochondrial degradation. Patients who lack FBXL4 have too few mitochondria in their tissues which leads to disease.
“Further studies are needed to explore the therapeutic potential of these findings, in particular whether inhibition of the degradation of mitochondria may provide a new treatment strategy,” says Nils-Göran Larsson.
The study was financed by several bodies, including the Swedish Research Council, the Knut and Alice Wallenberg Foundation, the European Research Council, the Swedish Cancer Society, and the ALF agreement between the Swedish government and the regional councils.
Publication
”FBXL4 deficiency increases mitochondrial removal by autophagy”. David Alsina, Oleksandr Lytovchenko, Aleksandra Schab, Ilian Atanassov, Florian Schober, Min Jiang, Camilla Koolmeister, Anna Wedell, Robert W. Taylor, Anna Wredenberg, Nils-Göran Larsson. EMBO Molecular Medicine, online 11 June 2020, doi: 10.15252/emmm.201911659.